

2026.01.11
818号令的核心突破在于构建了适配生物医学新技术特性的监管框架,彻底改变了以往单一化的审批模式,形成“备案制”+“审批制”的制度设计。
1.核心监管原则明确
条例确立“以人民健康为中心、防风险保安全、支持转化应用、强化责任落实”四大原则,首次在行政法规层面实现“鼓励创新”与“防控风险”的制度性平衡。这种平衡并非简单折中,而是通过“备案”提升创新活力,通过“审批”守住安全底线,为真正具备技术实力和合规意识的主体开辟“安全赛道”。
2.两阶段管理体系落地
临床研究阶段:实行“备案制”,经非临床研究证明安全有效后,需通过学术审查和伦理审查,在5个工作日内向国家卫健委备案即可开展,大幅缩短创新技术进入临床研究的周期。
临床转化阶段:实行“审批制”,临床研究需充分证明技术安全有效且符合伦理原则,经国家卫健委批准后方可转化应用,获批后还需接受常态化再评估。
3.分类分级与边界厘清
条例明确将生物医学新技术界定为“作用于人体细胞、分子水平,尚未临床应用的医学手段”,涵盖细胞治疗、基因治疗、再生医学等前沿领域。同时明确将制定技术与药品、医疗器械的界定指导原则,厘清“医疗技术”与“药品”的监管边界,避免双重监管或监管真空。
818号令通过刚性条款划定了行业合规底线,对临床研究、转化应用、风险防控等关键环节提出明确要求,违规成本显著提升。
1.临床研究的资质与流程硬约束
实施机构仅限三级甲等医疗机构,需具备专业伦理委员会、学术委员会及配套设施、专业人员和稳定经费。
禁止非医疗机构、非专业人员开展相关诊疗,法律明令禁止或存在重大伦理问题的技术(如生殖目的人类克隆)不得开展研究。
研究方案发生实质性变更(涉及研究目的、方法、受试者等)需重新备案,未按要求备案将承担相应法律责任。

2.全生命周期数据与样本追溯
临床研究记录和原始材料需保存30年,涉及子代的需永久保存,严禁伪造、篡改、隐匿数据。
生物样本从采集、运输、存储到使用的全流程需建立可追溯链条,确保每一个环节都有据可查。
需建立不良事件报告与应急处置流程,发生严重不良反应需立即暂停研究,由伦理委员会评估是否继续。
3.受试者权益保护的刚性要求
必须取得受试者或其监护人的书面知情同意,方案变更影响受试者权益的需重新获取同意。
开展临床研究不得向受试者收取相关费用,若造成健康损害需及时救治,费用由发起机构或过错方承担。
鼓励购买商业保险,为受试者提供更全面的权益保障。
4.法律责任的严厉惩戒
条例实行“双罚制”,对违规机构最高可处500万元罚款或违法所得10倍罚款,对相关责任人可处终身禁业。违规情形包括未备案开展研究、虚构材料、未履行伦理审查义务、损害受试者权益等,形成强有力的监管威慑。
818号令的核心创新在于构建了“药品注册”与“医疗技术转化”并行的双轨体系,为不同类型的生物医学新技术提供了差异化的合规路径。
1.两条路径的核心差异
药品注册:监管主体为国家药品监督管理局(NMPA),核心是“药品”监管,需满足严格的CMC(化学、制造和控制)标准,通过大规模RCT临床试验证明安全性和有效性,获批后获得药品注册证书,以产品销售模式实现商业化。
医疗技术转化:监管主体为国家卫生健康委员会(NHC),核心是“医疗服务”监管,适配操作复杂、难以标准化的技术,通过高质量临床研究证明安全有效后,获批成为医疗服务项目,以技术服务模式实现商业化。
2.路径选择的关键考量
技术特性:标准化程度高、可大规模生产的技术更适配NMPA路径;个性化强、依赖临床操作流程的技术更适配NHC路径。
商业化需求:追求快速落地、优化现金流的企业可优先考虑NHC路径;旨在构建规模化产品矩阵的企业可选择NMPA路径。
资源配置:NHC路径需深度绑定三甲医院及行业KOL,共建研发平台;NMPA路径需投入巨资建设GMP生产设施,构建市场化销售网络。
818号令的实施将引发行业深度洗牌,不同市场主体需根据自身定位调整战略,同时关注配套政策的落地进度。
1.对各类市场主体的影响
三甲医院:成为临床研究核心枢纽,需强化学术与伦理审查能力,建立全流程管理体系,同时面临优质临床资源争夺加剧的局面。
Biotech企业:需尽快完成管线战略评估,明确路径选择,深化与三甲医院的战略合作,建立符合路径要求的合规体系。
CDMO/CRO行业:传统生产服务需求承压,但“备案咨询”“合规辅导”等新增需求开辟增长空间,行业面临结构性调整。
2.配套政策的关键衔接点
技术界定指导原则:将明确“技术”与“药品”的划分标准,是企业选择合规路径的核心依据。
在线服务系统:将实现备案、审批、信息报告等全流程线上办理,提升监管效率与透明度。
分类分级标准:将依据风险程度和伦理敏感度,对不同技术实行差异化监管,高风险技术将面临更严格的审查。
面对818号令构建的全新监管体系,凌生科技聚焦生物样本管理与细胞冻存的核心环节,推出高适配的合规支撑方案,为行业伙伴提供实战性保障。
在生物样本合规管理方面,方案严格匹配条例对样本全流程追溯的要求,搭建覆盖采集、运输、存储、使用的合规化模块,满足法规对数据留存与不可篡改的要求;同时提供备案材料整理、伦理审查辅助等服务,助力合作机构快速通过伦理审查与备案流程。
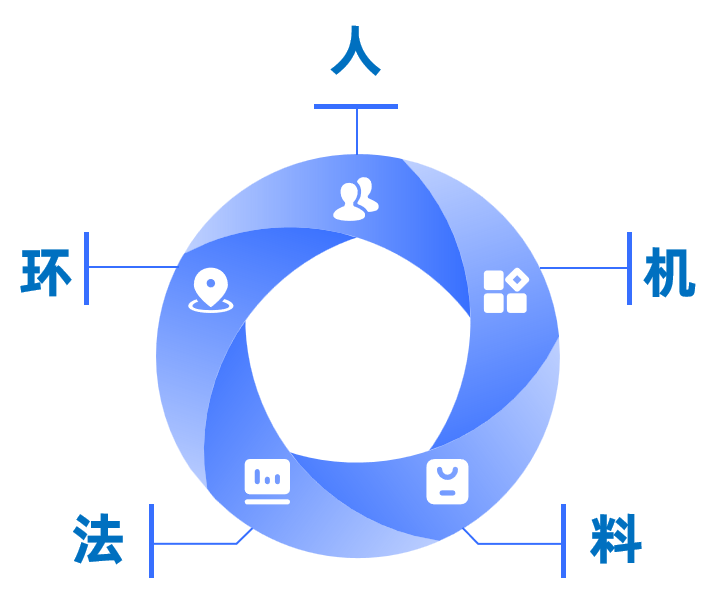
在细胞冻存液产品方面,采用无血清、无动物源成分配方,经严格无菌检测与毒性评估,符合临床研究安全性要求;生产过程遵循GMP规范,每批次提供完整质量检测报告(COA),确保细胞在深低温保藏期间的生物学活性稳定,为临床研究数据的可靠性奠定基础。
凌生科技的方案不局限于满足政策底线要求,更致力于通过标准化流程与高品质产品,帮助客户降低合规成本、提升研究效率,在监管重构的浪潮中抢占发展先机。



